Thyroid hormone (TH) receptors are present in the myocardium and vascular tissue, and minor alterations in TH concentration can affect cardiovascular (CV) physiology. The potential mechanisms that link CV disease with thyroid dysfunction are endothelial dysfunction, changes in blood pressure, myocardial systolic and diastolic dysfunction, and dyslipidemia. In addition, cardiac disease itself may lead to alterations in TH concentrations (notably, low triiodothyronine syndrome) that are associated with higher morbidity and mortality. Experimental data and small clinical trials have suggested a beneficial role of TH in ameliorating CV disease. The aim of this review is to provide clinicians dealing with CV conditions with an overview of the current knowledge of TH perturbations in CV disease.
The eagle-eyed among you might notice that the picture accompanying this post (taken from the actual paper) does NOT mentioned TRH. Which is the subject of another of my very recent posts:
Thanks Helvella, cardiologists appear to be much more ahead of the game than endocrinologists, who seem to have closed their minds to possibilities and think they have it all licked.
One of my frequent posts - and bought the book ! All research papers from when Cardiologists and Endocrinologists came together in Italy for the purposes of research back in 2004
How 'readable' is this book, Marz? Brain fog is making it hard for me to concentrate. I'm tempted to buy it but am wondering if it will be another one I can't take in a word of if it's too complex....
It is just Research Papers - so yes a hard read. I just read the introduction and the conclusion of most Research Papers
If you Look Inside on-line - you can read the Contents pages and see what they contain. Liothyronine - T3 - seems to be the star of the show ! If I lived in the UK I would lend you my copy ....
ah, thank you! I could just about stretch to the £35 for a used copy... but I seem to be collecting quite a stash of books I can't concentrate on...! I did take a peek at the look inside and it didn't seem tooooo unintelligble lol. Will have a thunk and consult my wallet!
My lipids consultant is rubbish, wouldn't acknowledge any link at all between low levels of T3 and heart disease. Bah! Good to hear some consultants are clued up though
Author(s): Avais Jabbar [1, 2]; Alessandro Pingitore [3]; Simon H. S. Pearce [1, 4]; Azfar Zaman [1, 2]; Giorgio Iervasi [3]; Salman Razvi (corresponding author) [1, 5]
Cardiovascular disease is a model of chronic degenerative disease, and at present is the leading cause of death worldwide, accounting for >15 million deaths each year [1]. According to 2020 WHO projections, cardiovascular diseases and their complications, in particular postischaemic heart failure (HF), will be the most important cause of morbidity and death worldwide, with high costs to health-care systems [2]. These forecasts reinforce the need to develop new therapeutic and preventive strategies to reduce cardiovascular disease morbidity and mortality [3].
Although standard risk factors and secondary prevention strategies for cardiovascular disease are well documented and firmly established, the role of other mediators in initiating and exacerbating underlying cardiovascular disease is increasingly acknowledged. The role of thyroid hormones -- particularly when abnormal -- in aggravating cardiovascular disease is recognized, given that thyroid hormone receptors are present in both myocardial and vascular endothelial tissues [4], thereby enabling changes in circulating thyroid hormone concentrations to modulate end-organ activity. However, the clinical significance of mild thyroid overactivity (subclinical hyperthyroidism) and underactivity (subclinical hypothyroidism) is uncertain. Moreover, the absence of high-quality clinical trials has led to controversy over the optimal protocols for diagnosing subclinical thyroid diseases and whether treatment of patients with mildly abnormal serum-thyroid results is appropriate. From a clinical perspective, 'subclinical' denotes the presence of disease without manifest symptoms and suggests the presence of either mild or early disease. Importantly, thyroid function tests are performed very frequently; for example, 25% of people in the UK have their thyroid function assessed annually [5]. Frequent testing can lead to detection of incidentally abnormal thyroid function parameters and, in the absence of high-quality evidence (either for or against treatment), can generate confusion as to the best evidence-based management of subclinical thyroid diseases.
Minor changes in circulating thyroid hormone concentrations can adversely affect the cardiovascular system, as evidenced by findings from observational studies showing that both subclinical hypothyroidism and subclinical hyperthyroidism are associated with a 20-80% increase in vascular morbidity and mortality [6, 7, 8, 9, 10]. These findings indicate that thyroid hormone status has great importance as a vascular risk factor in the general population, because [greater than equal ro]10% of women aged >55 years have subclinical hypothyroidism [6, 11] and 1-2% have subclinical hyperthyroidism [10, 12, 13]. Unfortunately, the role of subclinical thyroid disease as a cardiovascular risk factor is underrecognized, owing to the lack of high-quality outcome trial data to guide practice after the abnormal thyroid function is identified, and because of the lack of commercial exploitability.
This Review is intended for health-care professionals and researchers who are involved in caring for patients with cardiovascular disease or are studying potential treatments to ameliorate the complications of these conditions. The aim of this Review is to inform and update readers on the influence of thyroid hormones in the pathogenesis and the management of cardiovascular disease, and to summarize the current literature describing the effect of thyroid hormones in HF and acute myocardial infarction (MI).
Historical perspective
The association between thyroid dysfunction and cardiovascular disease is not new. In 1878, decades before thyroid function tests became available, William Greenfield described the autopsy findings of a middle-aged woman with myxoedema -- a severe form of hypothyroidism -- as having "much serous effusion in the pericardium ... the heart was large ... the arteries were everywhere thickened, the larger ones atheromatous" (REF 14). Soon after, clinical and autopsy accounts describing atherosclerosis in patients with myxoedema started to be reported [15, 16], and it became apparent that the clinical condition of myxoedema was linked to a poorly functioning thyroid gland. However, the diagnosis of myxoedema was based purely on clinical grounds and, in the absence of modern thyroid function assays, milder and less clinically apparent forms of the disease would have been missed. In 1891, George Murray subcutaneously injected sheep thyroid hormone extract into a patient with clinical features of myxoedema, producing dramatic improvement in symptoms and facial appearance [17]. Over the subsequent 30 years, Murray switched to oral thyroid extract collected from a pool of sheep, the glands being obtained from an abattoir.
Commercial production of a synthetic thyroid hormone (levothyroxine) took many years. This thyroxine was not available until 1949 and, even then, tablets of desiccated thyroid extract were the mainstay of replacement for many years [18]. Clinical trials in patients with myxoedema showed that this new therapeutic agent had positive effects in reducing high cholesterol levels [19] and reducing cardiovascular morbidity [20] and mortality [21]. From 1965 onwards, the first generation of radioimmunoassays to estimate serum thyroid-stimulating hormone (TSH; also known as thyrotropin) were developed, although these assays had limited functional sensitivity [22]). In 1967, in a case-control study including 25 autopsies from inadequately-treated patients with hypothyroidism and 50 age-matched and sex-matched control autopsies, Belgian researchers reported that the presence and severity of coronary artery disease was more common in patients with hypothyroidism (96%) than in euthyroid individuals (60%), and that left ventricular hypertrophy and dilatation was more frequent in the group with hypothyroidism [23, 24]. In 1977, Tunbridge et al . performed the first cross-sectional, cohort study in Whickham, UK, and concluded that no association existed between subclinical hypothyroidism and presence of cardiovascular disease, although a weak relationship was seen in women with subclinical hypothyroidism and minor electrocardiogram changes [25].
These initial data suggesting a possible link between mild or inadequately treated hypothyroidism and cardiovascular disease, as well as the benefits of levothyroxine in reducing cholesterol and cardiac morbidity and mortality, led investigators to consider the use of thyroid hormone analogues in euthyroid individuals at high risk of cardiovascular disease, with the aim of reducing the deleterious effects of thyroxine excess seen with levothyroxine therapy, while preserving its recognized beneficial effects on cardiovascular risk factors [26, 27]. The negative results of a trial of dextrothyroxine -- a low-activity dextro-isomer of thyroxine -- in male survivors of acute MI [28] curbed the enthusiasm for using thyroid hormone therapy in patients with cardiovascular disease. This trial showed increased arrhythmias and higher mortality in the treated group compared with untreated patients, probably owing to contamination with levothyroxine [28] and the supraphysiological doses of dextrothyroxine used in the trial (more than twice the normal endogenous thyroxine production) [29]. Another trial on the thyroid hormone analogue diiodothyropropionic acid in patients with HF showed higher heart rate and symptoms suggestive of hyperthyroidism in the treated patients [27, 30]. Of note, thyroid hormone analogues were used in both studies rather than native thyroid hormones. The results of these early trials prejudiced clinicians and curbed enthusiasm of researchers and pharmaceutical companies in developing and researching the use of thyroid hormones and its analogues at therapeutic doses in patients with cardiovascular disease. In the past decade, studies on the use of the liver-selective, thyroid hormone receptor agonist eprotirome in patients with resistant and familial hypercholesterolaemia showed a beneficial effect on blood lipid parameters [31], but this therapy has not progressed to large-scale, phase III studies owing to concerns over potential liver and cartilage injury [32].
Thyroid hormone action on the heart
The thyroid gland secretes two main iodinated hormones, T3 (3,5,3'-triiodothyronine) and T4 (3,5,3',5'- tetraiodothyronine; also known as thyroxine). Both molecules can generate biological activity in responsive tissues by binding to the thyroid hormone receptors; however, T3 is considered the biologically active hormone. The affinity of the thyroid hormone receptors is approximately tenfold higher for T3 than for T4 (Ref. 33); therefore, T4 must be converted to T3 to produce potent thyroid hormone receptor-mediated effects. Although T 4 is a prohormone for T3 , T4 can act directly through thyroid hormone receptors in a variety of tissues, such as blood vessels. For example, T4 can interact with the plasma membrane integrin [alpha]v [beta]3 , indicating that T4 is directly proangiogenic [34].
The thyroid gland secretes <20% of circulating T3 , with the bulk of T3 being produced from T4 in extrathyroidal tissues by a process of deiodination of a single iodine atom ([Box 1]). The most relevant pathway of thyroid hormone metabolism is regulated by three selenocysteine enzymes called deiodinases [35]. Two deiodinase enzymes -- type I iodothyronine deiodinase (DIO1) and type II iodothyronine deiodinase (DIO2) -- lead to extrathyroidal T3 production. DIO1 is active mostly in the liver and the kidney, and produces 15-20% of total circulating T3 (Ref. 35). DIO2 activity is located in brown adipose tissue, pituitary gland, brain, and heart, and is responsible for the majority (two-thirds) of T3 production [36, 37]. The third deiodinase, thyroxine 5-deiodinase (DIO3), catabolizes both T3 and T4 to inactive products, leading to the termination of thyroid-hormone action. Thyroid hormone status of an organism is dependent on both serum thyroid hormone levels and intracellular tissue levels, which are regulated by deiodinases. For example, increased DIO3 activity in cardiomyocytes decreases the levels of T3 in left ventricular tissue, causing a local hypothyroid state [38].
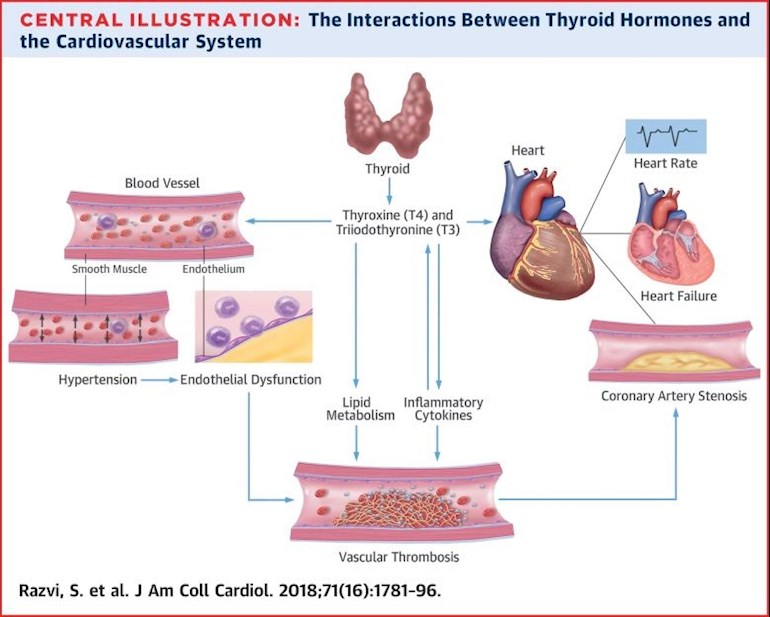
Thyroid hormones have a broad range of effects on the cardiovascular system, particularly on the heart (Fig. 1). Thyroid hormones influence cardiac status in three ways: by direct genomic actions on cardiomyocytes through binding to nuclear receptors, which leads to the regulation of the expression of target genes; by extranuclear, nongenomic actions on the ion channels in the cardiomyocyte cell membrane; and through the effects of T3 and T4 on the peripheral circulation, which determines cardiovascular haemodynamics, cardiac filling, and systolic contractility [4, 39].
In the cardiomyocyte, T3 binds to thyroid hormone receptors in the nucleus, which in turn bind to thyroid hormone response elements in the regulatory regions of target genes to regulate transcription. The two main thyroid receptors are thyroid hormone receptor-[alpha], which is highly expressed in cardiomyocytes [40], and thyroid hormone receptor-[beta]. Thyroid hormone receptors are unique in that they can bind to thyroid hormone response elements in the absence of thyroid hormones, leading to repression of transcription of target genes [41]. Therefore, the regulation of essential genes in the cardiomyocyte is dependent on the availability of thyroid hormones [42].
Thyroid hormone activity in the cardiomyocyte regulates myocardial contractility and systolic function. Thyroid hormones activate the expression of genes encoding sodium/potassium-transporting ATPases, myosin heavy chain-[alpha] (myosin 6; encoded by MYH6 ), and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2; encoded by ATP2A2 ), and negatively regulate the transcription of myosin heavy chain-[beta] (myosin 7; encoded by MYH7 ) and phospholamban (PLN ) [43, 44, 45] (Fig. 1). The two myosin heavy chains are important components of the contractile apparatus of the cardiomyocyte [46]. SERCA2 and its inhibitor PLN regulate the reuptake and release of calcium from the sarcoplasmic reticulum, thereby regulating the amount of calcium available for systolic contraction, which can determine diastolic relaxation of the heart. Thyroid hormones, by inducing increased levels of SERCA2 and decreased levels of PLN in the sarcoplasmic reticulum, promote the reuptake of calcium during diastole, leading to improved ventricular relaxation [47, 48, 49, 50]. Thyroid hormones also have a direct inotropic effect on the heart by positively regulating the gene expression of the [beta]1 -adrenergic receptor [51].
In addition, thyroid hormones influence cardiac chronotropy through both genomic and nongenomic effects on components of the adrenergic-receptor complex and on sodium, potassium, and calcium channels [4]. The effect of thyroid hormones on cardiac chronotropy manifests as tachycardia and increased risk of atrial fibrillation (AF) in hyperthyroid states, and as bradycardia and reduced cardiac contractility in hypothyroidism [4].
Nongenomic effects of thyroid hormones on cardiomyocytes and the systemic vasculature include activation of sodium, potassium, and calcium membrane ion channels [4], effects on the mitochondrial membrane and mitochondriogenesis [41], and involvement in signalling pathways of cardiomyocytes and vascular smooth muscle cells [48]. Thyroid hormones activate phosphatidylinositol 3-kinase (PI3K) / serine/threonine-protein kinase (AKT) signalling pathways, inducing production of endothelial nitric oxide and a subsequent reduction in the systemic vascular resistance [52]. Thyroid hormones influence cardiac mitochondrial function [53], and changes in the circulating levels of thyroid hormones might lead to impaired myocardial bioenergetic status and function [54]. Other nongenomic actions of thyroid hormones include vasodilatation [55]. Thyroid hormones decrease systemic vascular resistance by increasing production of nitric oxide and by increasing calcium reuptake within the arterioles, which leads to smooth muscle relaxation [55]. This decrease in systemic vascular resistance is also a result of the direct repression of PLN expression and the increase in tissue metabolism and thermogenesis induced by thyroid hormones [56]. The decrease in systemic vascular resistance induced by thyroid hormones, together with their direct inotropic effects, lead to an increase in cardiac output, as has been shown in patients with hyperthyroidism [57] and also after T3 infusion in high-risk patients undergoing CABG surgery [58].
The renin-angiotensin-aldosterone system also has an important role in the haemodynamic effects of thyroid hormones. The initial decrease in systemic vascular resistance induced by thyroid hormones leads to decreased perfusion in the kidneys, which increases renin and aldosterone levels [59]. The activation of the renin-angiotensin-aldosterone axis leads to an increase in the plasma volume and, therefore, an increase in cardiac preload, which is another explanation for the increase in cardiac output induced by thyroid hormones [4].
Overt and subclinical hyperthyroidism
Overt hyperthyroidism is commonly caused by stimulation of the TSH receptor by autoantibodies (Graves disease) or as a result of autonomous production of thyroid hormones by thyroid nodules [39] ([Box 2]; Table 1). The prevalence of overt hyperthyroidism in the general population is 0.5% [39].
Tachycardia is a common sign of overt hyperthyroidism, and 5-15% of patients with overt hyperthyroidism present with AF [4]. Normalization of thyroid hormone levels leads to the reversion to normal sinus rhythm in approximately 60% of patients who have AF owing to hyperthyroidism [60]. Shortness of breath during minimal exertion is also commonly reported by patients with hyperthyroidism [61]; however, the aetiology of this symptom has not been clearly defined. Moreover, hyperdynamic circulation -- characterised by increased preload and contractility, reduced systemic vascular resistance, and high heart rate, leading to a 50-300% increase in cardiac output -- is common in overt hyperthyroidism [39]. If overt hyperthyroidism is left untreated, or in those individuals with severe longstanding hyperthyroidism, this increased cardiac output can lead to symptoms and signs of HF as a result of left ventricular hypertrophy, arrhythmias (such as AF), and an increase in cardiac preload secondary to fluid overload [61]. Observational studies show that patients who have had an episode of hyperthyroidism have high long-term cardiovascular mortality, unless they received radioiodine therapy to induce overt hypothyroidism [62].
Hyperthyroidism also has a role in the pathogenesis of pulmonary hypertension [63, 64]. Epidemiological evidence supports a strong relationship between hyperthyroidism, pulmonary arterial hypertension, and right ventricular HF, with two studies showing a 43% and 44% prevalence of pulmonary arterial hypertension in patients with hyperthyroidism [63, 64]. Whether this relationship is incidental or thyroid hormones directly induce such a state is unclear. One potential mechanism is left ventricular HF and a hyperdynamic circulation secondary to an excess of thyroid hormones, which leads to an increase in pulmonary arterial pressure [61]. Studies in animal models have also shown that thyroid hormones cause endothelial cell proliferation and angiogenesis in pulmonary hypertension by binding to integrin [alpha] v [beta]3 and fibroblast growth factor receptors [65, 66]. These changes reversed after thyroidectomy [65, 66]. Few clinical studies exist on the effects of treatment of hyperthyroidism on pulmonary hypertension. The largest study included 64 patients with newly diagnosed Graves disease, of whom 28 had pulmonary arterial hypertension [64]. Follow-up echocardiography after treatment of hyperthyroidism in these patients showed normalization of pulmonary pressures in all patients except one [64].
Subclinical hyperthyroidism is defined by low circulating TSH levels with serum concentrations of T3 and T4 within the reference range [67, 68, 69, 70] ([Box 2]). Subclinical hyperthyroidism can be caused by exogenous (for example, secondary to excessive thyroid hormone replacement therapy or use of other drugs such as high-dose glucocorticoids) or endogenous (such as an underlying thyroid disease causing thyroid overactivity) factors. A substantial proportion (15-20%) of patients prescribed levothyroxine have a low TSH level, indicating exogenous subclinical hyperthyroidism [11, 71, 72, 73]. By contrast, the prevalence in the general population of endogenous subclinical hyperthyroidism depends on age, sex, and iodine intake, with a reported prevalence of 0.6-1.8% in iodine-sufficient areas in Iceland and the USA, and as high as 9.8% in iodine-deficient areas in Denmark [10, 12, 74]. Whether exogenous and endogenous subclinical hyperthyroidism are equivalent in terms of cardiovascular effects or the risk of cardiovascular disease is currently unclear.
Small, case-control studies including predominantly young patients (aged <60 years) with subclinical hyperthyroidism showed a higher heart rate, increased frequency of atrial and ventricular premature beats, and greater left ventricular mass compared with euthyroid individuals [75, 76]. However, this finding was not confirmed in larger, population-cohort studies in older individuals (aged >50-70 years) [77, 78]. Carotid intima-media thickness was also shown to be higher in patients with subclinical hyperthyroidism than in euthyroid individuals and patients with hypothyroidism [79]. A population-based study showed that low serum TSH is an independent risk factor for increased plasma levels of fibrinogen, which in turn have been associated with an elevated risk of cardiovascular events [80]. These risk factors would be expected to lead to an elevated risk of cardiovascular disease in patients with subclinical hyperthyroidism and, in keeping with this hypothesis, several observational studies showed an association between endogenous subclinical hyperthyroidism and incident cardiovascular disease [9, 81], AF [10, 82, 83, 84, 85], and cardiac dysfunction [75, 86]. A patient-level meta-analysis of 10 prospective, cohort studies including a total of 52,674 participants confirmed a strong association between subclinical hyperthyroidism and adverse cardiovascular outcomes [85]. After adjustment for age and sex, subclinical hyperthyroidism was significantly associated with increased coronary heart disease mortality (HR 1.29, 95% CI 1.02-1.62), and with increased risk of coronary heart disease events (HR 1.21, 95% CI 0.99-1.46) and incident AF (HR 1.68, 95% CI 1.16-2.43) [85] compared with euthyroid states. These data suggest a substantial contribution of mild thyroid hormone excess to the risk of developing AF, with an attributable risk (the number of cases of a disease among exposed individuals that can be attributed to that exposure) of 6.2% in the general population and up to 41.5% in those individuals with subclinical hyperthyroidism [85]. Interestingly, several studies also showed a positive correlation between serum T4 levels -- even within the reference range -- and risk of AF [84, 87, 88, 89]. Furthermore, even in patients with exogenous subclinical hyperthyroidism owing to levothyroxine use, those with fully suppressed serum TSH levels have an excess of cardiovascular and dysrhythmia events [90].
A definitive conclusion on the relationship between subclinical hyperthyroidism and cardiac outcomes is hampered by the varied and inconsistent methodologies used in the observational studies. Population heterogeneity, the different TSH cut-off levels used to define subclinical hyperthyroidism, the covariates included in the analyses, and different cardiovascular disease definitions all serve to invalidate interstudy comparisons. In addition, interpretation of a low serum TSH is clouded because this biochemical finding is associated with many chronic inflammatory disorders -- similar to the sick euthyroid syndrome, where TSH levels are usually slightly low, often in conjunction with a low or low-normal serum levels of free T3 -- and is also highly prevalent in healthy individuals at old age (>70-75 years) [91]. Most importantly, in the absence of randomized, controlled trials to examine the effects of treating subclinical hyperthyroidism on clinically relevant outcomes, the published consensus statement [68] and management guidelines [69, 70, 92] must be interpreted as expert opinion and in the context of other health issues present in each patient. Nevertheless, treatment of subclinical hyperthyroidism is advocated in international guidelines to prevent long-term complications, particularly when serum TSH levels remain persistently fully suppressed (<0.10 mU/l) in the absence of other explanations (such as use of levothyroxine or opiates). The European Thyroid Association guidelines [92] advocate treatment in patients aged >65 years with TSH levels <0.10 mU/l to ameliorate the risk of cardiovascular events, fractures, or progression to overt hyperthyroidism. Treatment can also be considered in patients aged >65 years with mildly low TSH levels (0.10-0.39 mU/l), particularly if they have underlying cardiovascular disease or other comorbidities [92]. In patients aged <65 years with TSH levels <0.10 mU/l, treatment is indicated in those patients with cardiovascular risk factors or comorbidities, persistent thyroid disease, and/or symptoms of thyroid disease, especially if thyroid autoantibodies are present [92]. In practice, further evidence of a tendency to thyroid hormone excess, either through demonstration of actual thyroid disease (by antibody assays or thyroid imaging) or a serum free T3 at the higher end of the reference range (6-7 pmol/l), gives a more robust support to endorse treatment of subclinical hyperthyroidism.
Overt and subclinical hypothyroidism
Overt hypothyroidism is diagnosed when serum TSH is elevated (usually >10 mU/l) and circulating free T4 is low (<9-10 pmol/l) ([Box 3]). The prevalence of overt hypothyroidism in nonpregnant adults is 0.2-2.0% [11, 93]. The causes of hypothyroidism are outlined in Table 1.
Overt hypothyroidism has several cardiac manifestations, including a reduction in cardiac output and cardiac contractility, a decrease in heart rate, and an increase in peripheral vascular resistance [94]. Marked changes in modifiable atherosclerotic risk factors also accompany overt hypothyroidism, including hypercholesterolaemia, diastolic hypertension, increased carotid intima-media thickness, and reduced production of endothelial-derived relaxation factor (nitric oxide) [95]. All these clinical features are reversible with thyroid hormone replacement therapy [95].
Subclinical hypothyroidism is diagnosed when serum thyroid hormones are within the reference range, but serum TSH concentrations are elevated ([Box 3]). Subclinical hypothyroidism can be mild (TSH >4.0-4.5 mU/l, but <10.0 mU/l) or severe (TSH >10.0 mU/l). However, no consensus exists on the 'normal' upper range of serum TSH, leading to controversy on both the definition and the clinical significance of subclinical hypothyroidism [96]. Prevalence of subclinical hypothyroidism in the general, adult population is 4-20% [69]. This wide prevalence range is a result of differences in age, sex, body mass index, race, dietary iodine intake, and the cut-off concentrations of serum TSH that are used to define the condition. Prevalence of raised serum TSH concentrations is higher in white than in black populations [97]. Estimations indicate that at least 10% of old women (aged >60 years) have subclinical hypothyroidism [98]; this prevalence has potential relevance as a vascular risk factor in the wider population.
The most frequent cardiac abnormality in individuals with subclinical hypothyroidism is diastolic dysfunction owing to impaired ventricular filling and relaxation [99, 100]. Although studies investigating systolic dysfunction in individuals with subclinical hypothyroidism have yielded inconsistent results, a robust study demonstrated that subclinical hypothyroidism was associated with systolic dysfunction (assessed by cardiac MRI) that reversed with thyroxine replacement therapy [101]. The presence of subclinical hypothyroidism has also been associated with diastolic and systolic dysfunction during exercise, resulting in impaired exercise tolerance in these individuals [102, 103]. Subclinical hypothyroidism can impair relaxation of vascular smooth muscle cells [39], inducing increases in systemic vascular resistance and arterial stiffness, and can also induce changes in endothelial function by reducing nitric oxide availability, without apparent clinical implications [104]. These findings are supported by population studies, with the Whickham Survey cohort [8] revealing higher systolic and diastolic blood pressures and total cholesterol concentrations in individuals with subclinical hypothyroidism than in euthyroid individuals, and the EPIC-Norfolk study [105] showing that, despite a worse profile of risk of cardiovascular disease in patients with subclinical hypothyroidism, coronary heart disease and all-cause mortality did not increase in the 10.6 years of follow-up.
Evidence from population studies on the association between subclinical hypothyroidism and cardiovascular morbidity and mortality is conflicting. Some prospective, population-based, cohort studies showed an increased risk of cardiovascular disease and death in individuals with subclinical hypothyroidism [7, 8, 106], but these findings were not confirmed in other studies [105, 107]. Patient-level meta-analysis of several prospective, cohort studies (with a total of 542,494 person-years of follow-up) showed that subclinical hypothyroidism is associated with a higher risk of cardiovascular events and death in people with high serum TSH levels, particularly in those with TSH levels >10 mU/l (Ref. 108).
Evidence from observational studies suggests a link between subclinical hypothyroidism and adverse cardiovascular outcomes [109, 110]. However, no randomized clinical trials are available to show whether improvements in cardiovascular events occur with thyroid hormone treatment of subclinical hypothyroidism. Small, interventional trials of levothyroxine therapy in individuals with subclinical hypothyroidism reported improvements in left ventricular function, vascular endothelial function, and atherogenic lipid particles, as shown by surrogate markers [39, 69]. Several other studies in patients with subclinical hypothyroidism showed similar results of improved cardiac function with levothyroxine therapy [99, 100, 101]. In an observational study including 3,093 patients with raised serum TSH, patients aged <70 years who were treated with levothyroxine had fewer cardiovascular events than untreated patients [111]. However, in individuals aged >70 years (n = 1,642), levothyroxine treatment had no benefit [111]. Interestingly, a small, interventional trial in patients with subclinical hypothyroidism showed improvement in cardiac mitochondrial function with levothyroxine treatment [54]. This study provides a novel insight at the subcellular level of the action of thyroid hormones in cardiac tissue [54] (Fig. 2).
Owing to the lack of evidence from prospective, randomized, controlled trials in subclinical hypothyroidism, international guidelines indicate that treatment should be considered only in those individuals with severe disease (serum TSH >10 mU/l), with symptoms of hypothyroidism, or in individuals aged <70 years, particularly if they also have other cardiovascular risk factors [112].
Cardiovascular risk factors
Hyperlipidaemia. Thyroid hormones are involved in lipid metabolism [95]. Overt and subclinical hyperthyroidism do not adversely influence lipid parameters [113] (Table 2). By contrast, the association between overt hypothyroidism and hyperlipidaemia has been known for many years, with some estimates showing a link in up to 90% of patients with overt hypothyroidism [94, 114]. Elevated plasma lipid levels are also evident in some patients with subclinical hypothyroidism [115], suggesting an increased risk of atherosclerosis in these individuals. However, population-based studies have shown conflicting findings. In some studies, such as the Whickham [93] and NHANES III [116] surveys, no link was found between subclinical hypothyroidism and hyperlipidaemia. By contrast, other studies showed a positive association between increasing serum lipid concentrations and serum TSH levels [116, 117].
The causes of hyperlipidaemia in an underactive thyroid state are the decreased expression of hepatic LDL receptors [114] -- which reduces cholesterol clearance from the bloodstream -- and the reduced activity of cholesterol-[alpha]-monooxygenase [114, 118], an enzyme that breaks down cholesterol. Randomized, placebo-controlled trials designed to assess whether levothyroxine treatment in individuals with subclinical hypothyroidism has a beneficial effect on LDL and total cholesterol levels had heterogeneous results. Some of these trials have shown a beneficial effect of levothyroxine replacement therapy on lipid profiles [119, 120, 121], whereas other studies have not [122, 123]. Such differences are probably the result of different population groups, varying severity of hypothyroidism, the presence of thyroid antibodies, different doses of levothyroxine, and the age of the participants. Meta-analyses of clinical trials indicate that levothyroxine therapy leads to a modest reduction in serum total cholesterol levels (approximately 0.2 mmol/l) in suboptimally treated patients with hypothyroidism or subclinical hypothyroidism [124]. A Cochrane review including six randomized clinical trials that had an assessment of lipid parameters showed that levothyroxine treatment for subclinical hypothyroidism had no overall effects in reducing plasma levels of total cholesterol, HDL, or LDL [125]; although a subgroup analysis showed a trend towards reducing LDL levels in patients with serum LDL >155 mg/dl (Ref. 125). In two randomized clinical trials that were published after the Cochrane meta-analysis, t
Thank you. And I think you might just be due a prize for the longest single reply ever posted here.
Quoting from that:
Unfortunately, the role of subclinical thyroid disease as a cardiovascular risk factor is underrecognized, owing to the lack of high-quality outcome trial data to guide practice after the abnormal thyroid function is identified, and because of the lack of commercial exploitability.
Gosh, thanks for that. It’s going to take me a while to read it properly. When people say that if you have one condition (aka diagnosis) then you are more susceptible to have others, then you start wondering about the interrelationship. I’ve found it hard enough understanding the thyroid bits (TRH, TSH, T4, T3 and any I’ve missed). But this shows that it affects cardiovascular as well and mentions that there are T3 and T4 receptors in all parts if the body. No wonder blood pressure and cholesterol levels can be affected and in my case probably blood glucose as well!
So do we treat symptoms or diseases or a combination if both. Fascinating stuff. I can’t think why researchers aren’t interested just because they can’t see the immediate financial benefit....
Fascinating. I did not understand it all, but what I did understand was illuminating. I hope this might indicate a trend to take thyroid conditions more seriously.
He did a post once - Stop Treating Thyroid Patients like Children. Of course the Thyroid Heart connection would be great. I think he once commented that he needed to finish - What really causes heart disease - before embarking on the thyroid.
Content on HealthUnlocked does not replace the relationship between you and doctors or other healthcare professionals nor the advice you receive from them.
Never delay seeking advice or dialling emergency services because of something that you have read on HealthUnlocked.

")
