Patients discontinue BTKi's due to resistance and intolerance. We evaluated the safety and efficacy of Pirtobrutinib (working name; formerly known as LOXO-305), a highly selective, reversible BTK inhibitor, in these patients. The study started recruiting November 16, 2018 and follow up is expected to continue until February 2023.
This was a first-in-human, multicentre, open-label, phase 1/2 trial of the BTK inhibitor pirtobrutinib. The primary endpoint was the maximum tolerated dose (phase 1) and overall response rate (ORR; phase 2).
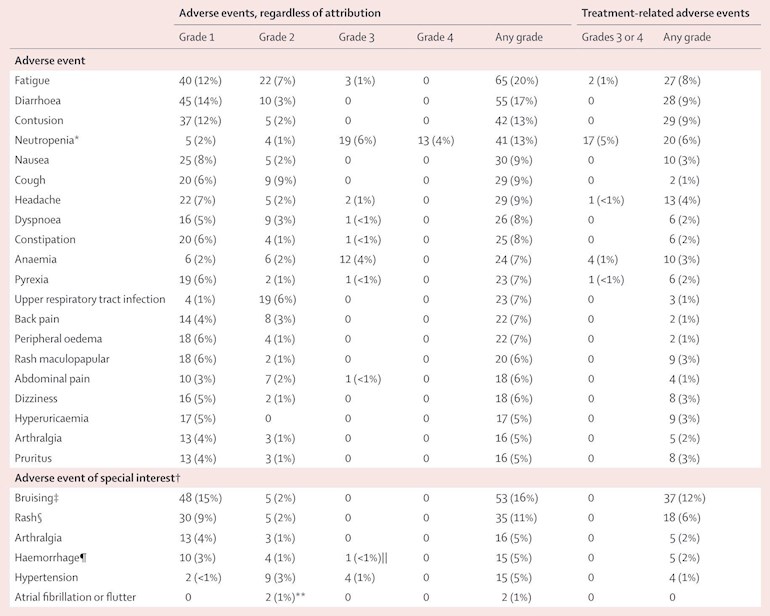
In 121 efficacy evaluable patients with CLL or SLL treated with a previous covalent BTK inhibitor (median previous lines of treatment 4), the ORR with pirtobrutinib was 62% . The ORR was similar in CLL patients with previous covalent BTK inhibitor resistance 67%, covalent BTK inhibitor intolerance 52%, BTK C481-mutant 71% and BTK wild-type 66% disease. Of 117 patients with CLL, SLL, or MCL who responded, all but eight remain progression-free to date Grade 3 atrial fibrillation or flutter was not observed.
Conclusion was that Pirtobrutinib was safe and active in multiple B-cell malignancies, including patients previously treated with covalent BTK inhibitors. Pirtobrutinib might address a growing unmet need for alternative therapies for these patients.
This is an important new BTKi for those that have developed resistance to Ibrutinib, particularly those with the BTK C481 mutation and offers a potential alternative to Venetoclax as a next treatment. The trial is ongoing and we look forward to seeing more data in the future.
Many thanks for sharing these encouraging updates. Can you share reasons why if/when available an Ibrutinib relapsed patient would opt for this rather than Venetoclax? Thanks.
If I relapsed because of a BTKi mutation that meant the Ibrutinib was ineffective I would definitely look at a trial of Pirtobrutinib and keep the Venetoclax in my back pocket for a later date, should I need it.
In the future it will probably be good to have testing for BTKi mutations before deciding the next treatment as I think there will be several BTKi's that can overcome various mutations.
It’s all about sequencing. The good news is there is another treatment in the arsenal the bad news is trying to determine which card to play next. It’s all very confusing.
I agree. Time will benefit us all as it relates to treatment sequencing. My problem is I go for a check up this week and now that the start of my BR treatment is 34 months in the rear view mirror I realize I’m on borrowed time. I’m not looking forward to making a second treatment selection. Hopefully, I will continue to be a positive outlier for a while longer. It’s always stressful facing a check up.
Jeff was just offering me encouragement. I said I was on borrowed time as the length of my remission from BR is starting to move beyond what the average unmutated patient experiences. He wrote he had received a five year remission from BR and he was doing well on Ibrutinib. It’s all good. I’m just lamenting about my upcoming appointment. It happens to the steadiest among us 😁.
Good response. I am 31 months post B+R as un-mutated. When I started B+R I was told that 36 to 42 months average for B+R. Mutated was 60 months average to relapse on B+R. My CLL was so aggressive that I do blood tests every 3 months, so far doing great. Blessings.
We are on the same path. I was diagnosed in October of 2017 and I started treatment in early May of 2018 for six consecutive months. I am also doing well. I started with quarterly appointments and the doctor pushed me back to every four months when Covid hit. My CLL was also nasty when it got going. After treatment I had residual disease in the marrow of 2 cells per 10,000. So that was a pretty good outcome. Let’s hope we both get at least a couple more years out of old BR.
That is my preference. I would ask for BTK mutation testing and then try to find a trial of a BTK inhibitor that was effective against my mutation. Of course, they may not find one or there may not be another effective BTKi and then I would be very happy to have Venetoclax.Jackie
Non-reversible or covalent bonding is a very strong bond. Reversible bonding is a weaker bond. An illustration is two ways of sticking a balloon to the ceiling. You could non-reversibly glue the balloons to the ceiling, or you could reversibly stick the balloon to the ceiling with static electricity. (Sometime, try rubbing a balloon on your hair or clothing, then placing it on the ceiling ).
The water molecule, H2O is a good example of a covalent/"irreversible" bond. It's very hard to split water back into hydrogen and oxygen. Water molecules also form weak/"reversible" bonds with other molecules, including other water molecules. That's why water is a "universal solvent".
Thanks Neil. It's more the biology I was trying to understand. Here's my broad take from a quick google. Please correct as necessary.
BTK is an enzyme which among other things acts as an oncogenic signalling molecule essential to the functions and survival of cancerous B cells.
Irreversible BTK inhibitors like Ibrutinib, Acalabrutinib, Zanubrutinib bind covalently to a specific amino acid C481 (C I believe stands for cysteine and 481 for its position in the peptide chain of 659 amino acids), thereby inhibiting BTK signalling and rendering the cancerous B cells inactive (although no longer proliferating they survive in a dormant state?).
The drug becomes ineffective if it can no longer bind to C481 because of a new mutation (I don't know if this is altogether random or is drug induced), the most common mutation being a straight substitution of serine for cysteine, i.e. C481S. We now have BTKi resistance.
Reversible BTK inhibitors are being developed, like Loxo305/ Pirtobrutinib, Remibrutinib et al. These bind to BTK non covalently and at a different binding site where it also inhibits the functions of BTK. Such drugs may also have a role for patients who have encountered resistance to BCL2 inhibitors like Venetoclax.
My understanding is that this mutation is usually drug induced. And that the BTK bond at that specific site becomes reversible instead of irreversible. The change in the amino acid (cysteine to serine) doesn't allow the molecule to form a strong enough bond that it can be considered "irreversible". The physical structure of the serine won't allow it. Imagine a small 6 sided box that can fit inside a slightly larger 4 sided box. The 6 sided box doesn't touch on all sides of the box like a slightly smaller 4 sided box would. And that different makes the difference between "irreversible" and "reversible" binding. For irreversable, we need both boxes to be the 4 sided match.
And the new reversible-binding drug works at a different place than C481 along the BTK enzyme. I want to clarify that, since it doesn't seem to be the fact of "reversible" versus "irreversible" binding that makes the new drug effective. Even though the "new" drug has a reversible binding, that amount is enough to distort the shape of the BTK enzyme in CLL cells so that it doesn't work as well, and the CLL cells die. And the binding of the new drug's "reversible binding" box is still strong enough to disrupt BTK. It's the shape disruption, not the reversible vs. irreversible. For anyone interested in stereochemistry 😂.
Why can't they crack the code to destroy all bad cells at once? Why do these pesky bad cells survive or hide? It seems to me that they have their own minds invading our bodies wherever they have the opportunity. I wish they would come up with something to melt them like "fire".
Well, until we find something unique to the CLL cells that other cells don't share, it's difficult. We know BTK is important in CLL, but unfortunately other cells also use it. BTK is a member of the Tec family tyrosine kinases and is expressed in all hematopoietic cells except T cells, natural killer cells and plasma cells. Cells generally become cancerous because 1) an outside agent indues a DNA deletion or mutation, and/or 2) DNA repair enzymes fail to properly repair DNA, especially before a cell replicates. Our DNA is "broken" numerous times throughout our life by things that also confer benefits--like ultraviolet radiation in sunlight. We make Vitamin D, but that energy also breaks DNA, which our bodies repair. As we age, our DNA repair enzymes may not work as well. As we make new cells containing DNA repair enzymes, an early failure to correctly copy that enzyme means we have a defective enzyme at an earlier age. Or perhaps a chemical or other reason (missing or defective gene from a parent?) causes a DNA issue. There doesn't seem to be a specific "cause" for a mutation or deletion that happens to occur on any part of a DNA strand (17p deleted area, or induces an extra copy in area 12 of a chromosome so we get Trisomy, or wherever). Like a tire can blow out for any number of reasons, with variables that affect whether or not a tire blows at that time, our DNA may or may not be affected by things as we live.
The general thinking is that CLL has many sub clones existing in the same patient at the same time.
For treatment naive patients who become resistant then they already have the resistant clone when treatment starts. The selective pressure of Ibruitnib means that all other clones are suppressed but the resistant clone slowly expands and causes relapse/resistant disease. This is a rare situation for naive patients, some of whom are now still in remission after nearly 9 years of treatment.
As BTKi's are effectively non proliferation drugs, it's harder for them to develop resistance and may account for the very long remissions that are seen.
For previously treated patients then previous treatments, especially chemo-immunotherapy, means they have often developed many mutations and clones which may have resistance to various therapies. Again the resistant clone grows and becomes dominant whilst the other clones are reduced by treatment.
Thanks for your explanation SofiaDeo, nice analogy.
I looked up cysteine and serine and noticed how similar they are structurally, just one atom different in the R group: S in cysteine versus O in serine is the extent of the mutation that differentiates covalent from non covalent binding and causes Ibrutinib resistance.
"Besides its role in BCR signaling, BTK also plays an role in signaling of cytokine receptors, CD19, CD38, CD40, chemokine receptors, such as CXCR42, tumor necrosis family receptors (TNFR), toll-like receptors (TLRs), and integrins. Of particular interest are effects of BTK on cell motility and tissue homing, given that the BTKi cause redistribution of tissue-resident (CLL) B cells into the peripheral blood, causing lymphocytosis that depends on the continuous presence of the BTK inhibitor3, 4. The role of BTK in chemokine receptor- and integrin-signaling in B cells2, 5 is considered to be the basis for this clinical phenomenon."
CLL cells aren't immortal and with the BCR "stay alive" stimulus blocked by the BTKi, which also disrupts CLL cell development, they eventually succumb to apoptosis in the peripheral blood, where they are also more vulnerable without their protective micro-environment.
SofiaDeo,
You are right that it is a change in the BTK structure that prevents covalent (irreversible) bonding, but it can't be drug induced, because unlike the older "chemo" drugs, BTK inhibitors do not affect the CLL DNA. It's just natural selection pressure that leads to any CLL cells not inhibited by the covalently bonded BTKi to become dominant. Per the aforementioned reference "CLL progression often coincides with an expansion of clones carrying BTK mutations at the ibrutinib binding site (C481S) or mutations in PLCG2 (R665W, L845F, S707Y), the gene encoding this BCR signaling-related molecule18, 61, 63. While BTK mutations generally reduce binding and thereby the efficacy of the kinase inhibitor, activating PLCG2 mutations result in pathway activation that is independent from BTK63. In addition, ibrutinib therapy can also promote the expansion of CLL sub-clones carrying del(8p), with additional driver mutations64. Based on mathematical models65 and verified with a highly sensitive droplet method for detection of single cells with somatic gene mutations64, it is apparent that in some patients miniscule populations of resistant cells can be already present before therapy initiation, which then become selected and expand under therapeutic pressure."
In this post, healthunlocked.com/cllsuppo... I've highlighted which BTKi drugs are covalent (irreversibly) or non-covalently (reversibly) bonded. The reference mentions a few of the new BTKi drugs I've listed and how they differ from Ibrutinib.
Sorry, I've been out all day. Everyone, including yourself, have explained it eloquently enough. I would only add that an irreversible, covalent binding bonds specifically to a target and this leads to a very selective binding which gives high potency to the drug.
Reversible binding drugs are in equilibrium with their target and are continually binding, unbinding and rebinding.
I wonder if the reversibles show less "side effects" because of this. They "unbind" enough to allow normal cell function, but "bind" enough to disrupt CLL cell function.
Yes, apparently so but the potency is then compromised and higher doses would be required for the same therapeutic effect. Can't win! The answer is more selective BTKi's and Acalabrutinib and Zanubrutinib show promise there with less off target side effects.
CRISPR gene editing. It already exists, but CLL has many different mutational causes. If you were able to target just one CLL stem cell progenitor, it would work.
Dr Mato said that because of the weaker bond loxo-305 isn’t always bonded to the B cell which (somehow) puts less pressure on the B cells to mutated around the drug. Or something like that.
Thanks for the link, I'll settle for Dr Mato's brief explanation for the time being. I tried to approach the question from the direction of Ibrutinib resistance. That just put me in my placeyoutu.be/pzk6hnaYP7s Dr Furman's third slide - yikes!
Here is a good write-up from MedPage Today about this promising treatment option. The article also links to an expert roundtable discussion and an editorial commenting on its advantages. More treatment options are definitely better, so add what looks to be a very good one to the treatment toolbox.
Pirtobrutinib Makes Splash as New CLL Drug
— Highly active in pretreated patients, even prior BTK inhibitor exposure, with little toxicity
by Ian Ingram, Deputy Managing Editor, MedPage Today March 8, 2021
This is a very pertinent video presentation (with transcript) from Dr R Furman, covering the range of BTKi progressives. He sees improved prognostic markers as the key to optimising treatment and avoiding some disease progression/ transformation.
One issue with Ibrutinib resistance is that sometimes a BTK mutation driving it was established during W&W, and treatment just selects for that mutant. Knowing this before a patient started treatment would be useful to say the least! oncnet.com/videos/treating-...
Thanks Jackie. Late response from me, my turn to be out all day.
Jan Burger at MDA, mentioned in the video, seems to have developed a high speed technique "Using droplet-microfluidic technology and growth kinetic analyses, we demonstrate the presence of ibrutinib-resistant subclones and estimate subclone size before treatment initiation" Full details of methods here nature.com/articles/ncomms1... most of which is gobbledegook to me. Maybe Dr Furman hopes that something like this can be used to routinely screen patients and guide treatment decisions?
Content on HealthUnlocked does not replace the relationship between you and doctors or other healthcare professionals nor the advice you receive from them.
Never delay seeking advice or dialling emergency services because of something that you have read on HealthUnlocked.

") ).
).